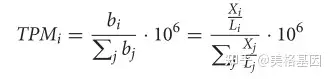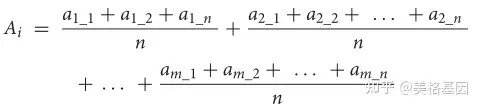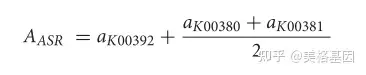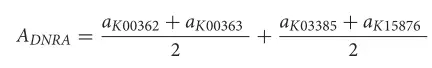|
- b. {* G x. p# x$ |" S/ v! l
Diting工具包是一个什么样的工具包呢,我们找到原文进行解读,一起给大家带来详细的介绍,关注美格基因公众号,后台回复0718即可获取原文。 : x# u7 w3 V) r1 d$ ^ Q6 j
9 C4 [$ l' V1 a! h4 ]
标题:Diting: A Pipeline to Infer and Compare Biogeochemical Pathways From Metagenomic and Metatranscriptomic Data
# {" l& w0 J' u" u7 b# c' A" h, ? 译名:Diting:从宏基因组和宏转录组数据推断和比较生物地球化学途径的管道 # R- z; }7 L- f/ g" f
作者:Chun-Xu Xue等 " B; S: E% i: X( T6 @! @. X- I
时间:2021-08-02
, d2 |* R3 h& ~4 X0 G9 p! A% K 期刊:Frontiers in Microbiology
6 A- N7 u5 P# X9 |0 _ DOI:10.3389/fmicb.2021.698286
$ x1 H' {" ^4 D5 y" y; D 一、摘要
8 {5 Y' p/ m L9 b/ W/ c b 宏基因组学和宏转录组学是揭示自然生态系统中驱动生物地球化学循环的关键微生物和过程的有力方法。很少看到有从宏基因组/宏转录组数据中专门描述生物地球化学途径的数据库,例如:二甲基巯基丙酸内盐(DMSP)代谢,这是一种丰富的有机硫化合物。此外,尚未建立一个用来从宏基因组和宏转录组数据中估计相对丰度和环境重要性的途径的公认标准化模型。这些限制影响了准确地将关键的微生物驱动的生物地球化学过程与环境条件的差异联系起来的能力。因此,迫切需要一种简单、专业的工具来推断和直观地比较生物地球化学过程的潜力,包括DMSP循环。为了解决这些问题,作者基于KEGG和人工创建的DMSP循环基因数据库,开发了一个工具包Diting,用于一步推断和比较给定宏基因组或宏转录组数据之间的生物地球化学途径。对100多条路径的相对丰度进行了精确和具体的计算。输出报告以文本和图形形式详细描述了生物地球化学途径的相对丰度。将Diting应用于模拟宏基因组数据,结果表明模拟基准基因组数据的遗传特征是一致的。随后,应用于热液喷口和Tara海洋项目的宏基因组和宏基因组数据,将Diting预测的功能剖面与环境条件变化相关联。Diting现在可以应用于更广泛的宏基因组和宏转录组数据集,它可以在https://github.com/xuechunxu/Diting上获取。 7 A4 {4 P1 e% w* ?9 u4 V% w9 P
二、材料和方法 % C& K' {( Z2 y/ g4 z% K: Q
1.Diting装配的主要工序
) ~ h* n. G7 `! |& B' X. _ Diting是用Python 3编写的,运行在Linux/UNIX平台上。运行该软件所需的先决条件在Diting GitHub中进行了描述。Diting可以通过Conda2安装。输入源是一组宏基因组/或宏转录组组criptomic干净读数,其中低质量的读数、引数和适配器序列事先已经被修剪过(图1)。然后,输入数据集由Megahit V1.1.2或Metaspades V3.12.0根据用户的规范使用汇编器的默认参数进行组装。与Megahit相比,Metaspades在恢复长连接方面表现得更好。它具有更高的装配质量指数,是高复杂宏基因组的推荐装配器。然而,MegaHit错误率低,具有很高的记忆性,是大型数据集的理想选择。可选地,用户也可以在运行Diting之前自行组装读取。Diting支持组装的连续和干净的读取一起作为输入。
5 L! S+ Q5 p8 b! ~* Y6 F( q 图1. 运行DiTing的主要步骤流程图2.基因预测与量化 : p$ s2 u7 Q8 g
Prodigal V2.6.3使用“-p meta”选项预测和翻译基因。为了确定每个基因的相对丰度,输入的宏基因组读数通过BWA-MEM (BWA v0.7.15,默认设置)与预测的基因(核苷酸)进行映射,以生成序列比对图(SAM)文件。未排序的SAM文件被用作pileup.sh(BBMAP v38.22)(Bushnell,2014年,默认参数)的输入,以计算每个基因或转录本的平均覆盖率。TPM方法用以下公式表示基因的相对丰度。 & }1 g- p4 p" a
% Y# d- t3 ]) f5 s 其中TPMi是基因I的相对丰度,Bi是基因I的拷贝数,Li是基因I的长度,Xi是基因I在样本中被检测到的次数(即对齐的次数),J是样本中的基因数。 ; x6 r" R. ^5 J) M' L8 c
3.基因注释 : ?! `/ x( S8 y- p5 ]9 I7 @
使用HMMER内实现的HMMSEarch对照KOFAM数据库[HMM Database of Kegg Ortherologs(Kos)]查询翻译后的蛋白质序列,这采用了灵敏和准确地检测远端同源物的方法。Kofam建议使用values3作为HMMSearch的cuto helect阈值,其中每个KO条目都有其独特的cuto helect阈值。Kofamkoala为蛋白质序列分配KOS数,其精确度与现有最好的KO分配工具相当。 对于被分配到多个KOS数的基因,所有相应的功能都与该基因相关联。 为了特异性地探索DMSP的分解代谢,人工收集了20个已证实的基因序列(DMSP裂解酶基因DDDD、DDDK、DDDL、DDDP、DDDQ、DDDY、DDDW、ALMA1;DMSP合成基因DSYB、DSYB、MMTN;DMSP去甲基化途径基因DMDA、DMDB、DMDC、DMDD;丙烯酰辅酶A水合酶ACUH、甲硫醇S-甲基酶MDDA、二甲基硫醚(DMS)单加氧酶DMOA、甲硫醇氧化酶MTO和DMSO还原酶DORA)。用一个带有基因相对丰度和注释的表来估计每个样本中大约100条生物地球化学途径的相对丰度。
1 o, R8 W" B+ t2 s3 `* u 4.标准化 . }: G6 k T0 R# A" ]! d2 F7 G
每个通道的公式是专门设计来根据定义估计通道的相对丰度的。
; Y: s0 P' O" t; L
* N( q! o: C! F% i y3 N 其中ai是i途径的相对丰度,am_n是每个样品中蛋白质m_n的相对丰度。M是完成I途径的可选途径之一,N是可选途径M中的蛋白质数。例如,将亚硫酸盐转化为硫化物的同化亚硫酸盐还原(ASR)有两种已知的可能途径 1)SIR蛋白(K00392)介导途径,和(2)Cysji蛋白(K00380+K00381)介导途径。因此,ASR途径的相对丰度由以下公式估算: 1)SIR蛋白(K00392)介导途径,和(2)Cysji蛋白(K00380+K00381)介导途径。因此,ASR途径的相对丰度由以下公式估算:
h P! `9 X+ f! I3 l) Z : _/ v' f/ {* W, A3 Q7 L
其中AASR是ASR途径的相对丰度,AKO是每个样本中KO的相对丰度。异化亚硝酸盐还原(DNRA)是一种将亚硝酸盐转化为氨的酶促反应,可通过以下两种酶促反应实现:(1)NIRBD蛋白(K00362+K00363)将亚硝酸盐转化为氨;
`8 ]; {) l" ]3 ]$ o! j (2)NRFAH蛋白(K03385+K15876)将亚硝酸盐转化为氨。因此,DNRA对氨的相对丰度由以下公式估算: 4 s1 Z. F* k5 R1 H
- M5 s% j" [# D' A2 P' `
其中ADNRA是DNRA途径的相对丰度,AKO是每个样本中KO的相对丰度。对于其他途径,使用了针对每个途径的定制公式。 : n, C- j# R4 j! b+ n) s8 V K
Diting在指定的输出目录中生成一个表。该表包含大约100个生物地球化学途径及其在每个输入样品中的相对丰度。另外还生成了每个样品中这些途径内相应的KO/基因的相对丰度表。为了提高可视化程度,最后绘制了热图和草图,用于比较不同样品中生物地球化学途径的相对丰度。输出还包含了一些重要的微生物前沿www.frontiersin.org4中间数据,如组装序列、基因序列和映射文件。
8 r6 e" }! D0 ]) K7 [0 M 三、主要结果 7 L' A1 k. Y6 Z$ k/ |! j
1. DiTing概况 4 V) x$ W* w! Z# Y
作者开发了一个新的宏基因组学/宏转录组分析管道DiTing,以推断和比较关键生物地球化学循环的基因和途径的丰度。DiTing由四个主要功能组成: 8 l; c( Y6 e' A+ t" w* E
(1)自动组装、开放阅读框架(ORF)预测、映射和基因注释; 9 A( _' J t. p
(2)人工创建和整理DMSP循环相关基因数据库;
2 a3 a4 `3 ]+ J7 [/ J (3)DMSP和其他生物地球化学途径计算生物地球化学相关途径和基因相对丰度的具体公式; # J: m7 {! z! v4 h7 z! I
(4)可视化结果比较两个输入样品之间的生物地球化学循环电位。
% }, n' O' h, Q2 @( |' q' I7 ? 这些特性使DiTing成为研究生物地球化学循环的灵活和多功能的工具包,或者仅仅作为处理宏基因组鸟枪测序数据的平台。此外,DiTing具有较高的速度,5个总大小约为 500 Gb 的样本(来自下面的热液喷口案例研究)用于评估速度。在Linux 版本4.15.0-20-generic 服务器(Ubuntu 18.04;CPU,Intel(R) Xeon(R) Gold 6140 CPU @ 2.30)上使用60个CPU线程,从读取到可视化的所有分析的总运行时间约为33小时GHz;内存,256 GB)。
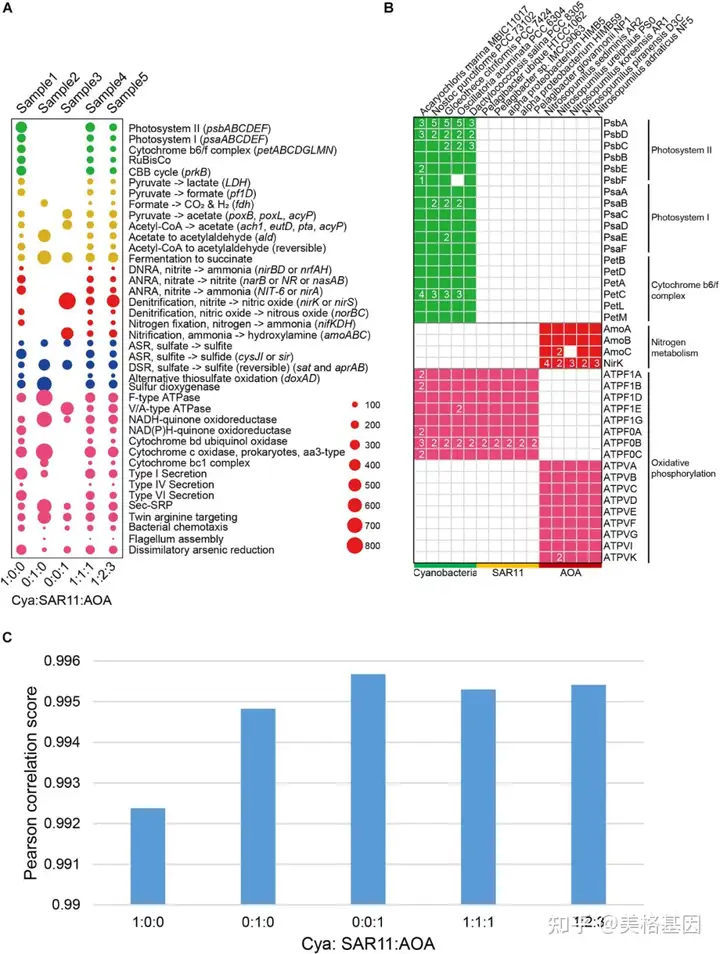
: d# S+ x# Y( Y9 z' I 2.用模拟基准数据集测试DiTing的精度
, S3 O) {8 d; z. p 为了验证DiTing的准确性,将DiTing在模拟数据上的结果与15个基因组的遗传特征进行了比较,并人工推断出KO家族相对丰度剖面(图2)。模拟样品中生物地球化学途径的总体相对丰度与模拟中使用的基因组的遗传特征一致(图2a、b)。随后,使用Kofamscan软件对来自15个基因组的用于模拟的翻译基因序列(氨基酸)进行注释。为了将DiTing得到的KO相对丰度剖面与真实KO相对丰度剖面进行比较,用PCC计算了这两个KO相对丰度剖面的相似性。所有PCC得分均高于0.99(图2c),表明DiTing建立的KO相对丰度剖面与实际结果有很强的一致性。以上结果验证了该方法的准确性。 " t$ g4 r; p+ n# J! N8 _
图2. 描述模拟宏基因组中通路相对丰度的DiTing结果的气泡图3.DiTing在五个真实热液喷口资料中的应用 . M* F! Q( p6 ]) L( e: B
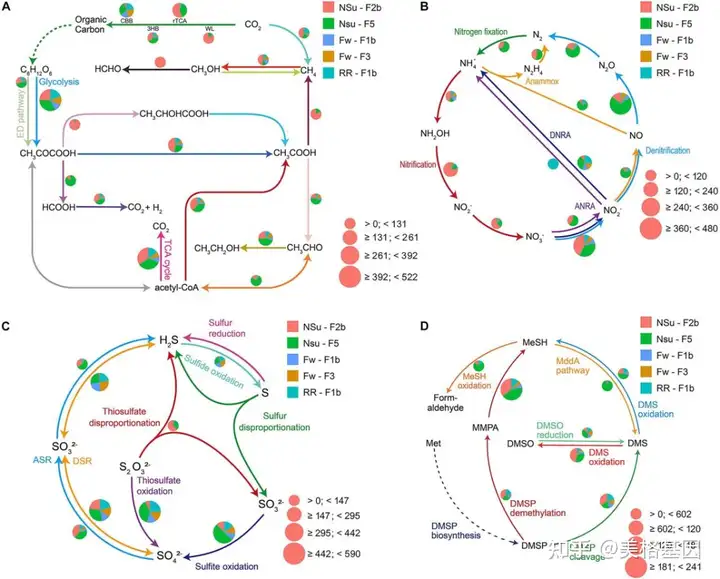
利用DiTing分析了从马努斯盆地Pacmanus和North Su油田采集的热液喷口样品生成的5个海洋宏基因组样品的生物地球化学潜力。每个样品的宏基因组clean reads范围从81到112 GBP。读取被组装成799,269到1,182,847个连续体,总组装尺寸从0.58到1.00GBP不等。然后预测了这些连续体中的总共5,639,558个ORF。大约18.9%的ORF在KEGG数据库中被注释,并且有8128个KO条目。根据新公式计算了大约100条生物地球化学相关途径的相对丰度。这些途径中的基因相对丰度也为进一步的基因水平分析做了准备。通过DiTing生成了这些途径可视化的概要草图(图3),这些草图反映了宏基因组样本中群落功能的直接模式。
1 a4 h6 _+ N. T; J 图3. 代表来自马努斯盆地的五个宏基因组样本的碳(A)、氮(B)、硫(C)和 DMSP (D)循环相关途径的相对丰度的饼图4.DiTing在15个真实TARA海洋工程数据集上的应用
) z6 I4 m9 }& j 应用DiTing方法对TARA海洋项目地中海叶绿素a(Chla)最大层的15个宏基因组样品进行了分析。每个样品的宏基因组clean reads范围为1.24~52.53GBP。读取被组装成71,183-1,601,956个连续体,总组装尺寸从0.045到1.38GBP不等。然后预测了这些连续体中的总共18,431,131个ORF。大约24%的ORF在KEGG数据库中被注释,并且有8759个KO条目。发现了与生物地球化学循环有关的74条途径。与来自样品的热液喷口相比,CHLA最大层包含了非常高的光系统路径的相对丰度。DMSP合成基因dsyb的相对丰度与原核DMSP合成基因dsyb的相对丰度相当,说明在这些环境中,原核生物和真核生物都产生了DMSP。对于DMSP降解,在15个样品中,有6个样品通过DMDA基因去甲基化DMSP的遗传潜力高于DMSP裂解(DDDS和ALMA1)(DMSP去甲基化:DMSP裂解=1.69:1)。这与热液喷口样本形成对比。在另外9个样品中,DMSP去甲基化的潜力与DMSP裂解的潜力相当(DMSP去甲基化 MSP裂解=0.82:1)。这些数据支持DMSP的去甲基化和裂解是CHLA最大层DMSP的主要分解途径。 MSP裂解=0.82:1)。这些数据支持DMSP的去甲基化和裂解是CHLA最大层DMSP的主要分解途径。
! j2 k" c1 g+ c 图4. 描绘碳(A)、硫(B)、氮(C)和其他选定(D)过程的路径相对丰度的气泡图5.DiTing在宏基因组数据集与宏转录组数据集组合中的应用 2 r; D" S; ?! O4 |# v+ L( j
随后,作者将DiTing应用于三个宏基因组样品及其相应的宏基因组。每个样品的宏基因组和宏转录组clean reads的大小分别为6.8~9.9GB和2.7~3.9GB。在Linux Version4.15.020-Generic服务器上使用60个CPU线程,所有分析从汇编到可视化的总运行时间为大约11小时。经过Diting的分析,生物地球化学途径的总体相对丰度与最初的研究一致(图5)。例如,CBB3型细胞色素C氧化酶基因/转录本在3个样本中被发现,但在标记113 2015变分样本中都不存在,根据DiTing输出和参考结果都不存在,CBB3型细胞色素C氧化酶基因/转录本。固氮酶铁蛋白(nifh)基因在一个宏基因组和两个宏基因组样本中均缺失。然而,参考研究与实验结果之间也存在一些不一致之处。这些NARG转录本可能在最初的研究中被遗漏,这是由于KO数据库中所有基因注释的通用阈值。相比之下,Diting根据Kofam的建议,为NARG使用了特定的cuto hust阈值(-domt 304.50),从而实现了正确的注释。 7 Q( I, J' q @4 ?' P
图5. 参考研究和DiTing之间宏基因组和宏转录组数据集组合的比较分析三、结论
) L5 U. b/ b8 D- s% L8 h 总之,本研究开发了一条管道(DiTing)来从宏基因组和宏转录组数据推断和比较生物地球化学途径。DiTing是一个用于分析宏基因组和宏转录组数据集的便携式工具,为数据处理提供自动的、多线程的生物信息工作流,包括读取组装、ORF预测、注释和用于计算生物地球化学路径相对丰度的定制特定公式。可视化模块旨在通过图形输出更容易地比较样本之间的功能。此外,人工建立了一个验证数据库,用于注释参与DMSP产生和循环的基因。作为对DiTing输出结果的验证,已发表的宏基因组中生物地球化学途径的相对丰度与DiTing计算的相对丰度的比较是一致的。应用DiTing对5个热液鸟枪宏基因组进行分析,结果表明,功能剖面能准确地反映环境条件(H2S和O2浓度)的变化。除海洋环境外,DiTing还可以很容易地应用于其他有趣的环境(如冰川、土壤环境和废水)。DiTing在相对简单的用户干预下可以很容易地应用于宏基因组和/或宏转录组研究。这一生物信息学框架将有助于我们理解微生物组介导的生物地球化学循环的时空变化。 2 W. J% q5 D5 x4 o) d9 K) T/ s$ k
您可能还喜欢:
# e: |; y& M# s% l! |
% i* t. }( ]6 I, N0 P2 v & o6 U: R( i' b- g
, a3 l' ]; R; M2 }. _
5 `4 p. ? r; C) Y+ S8 L* A4 c
! \0 g- m) W4 q4 Y9 r e3 o
% W8 f& J- x- e% y3 T* _, I, H% [4 l1 K) z/ f; |0 O% }9 j
|