|
6 U( c: y9 \" X+ ]: f6 g# ^ 撰文:黄朝 西北大学
, l/ T* c: Q K8 y 责编:刘永鑫 中科院遗传发育所 7 D+ H" N# g" x# K

4 @( U6 t# _ B' D' x4 K8 g$ | 淡水:21世纪的分子微生物生态学
6 [5 u) @+ ]4 u( I/ I8 O j 摘要- e3 x1 N- k% ^- ~
微生物无处不在,是具有生态意义的淡水环境分类和功能多样化的组成部分。淡水系统中的细菌,古细菌和真核微生物支持一系列生态过程和功能。然而,我们对微生物生态学知之甚少。随着人类新世纪的发展,对淡水的需求随着人口密度的增加而增加,并且越来越多地受到气候变化,富营养化和化学污染等多重且往往相互作用压力因素的威胁。因此,如果我们要有效地管理淡水环境的未来,就必须了解微生物的生态学及其在淡水生态系统中的功能作用。为此,研究人员讨论了微生物生态学分子实验技术的历史,并强调了这些方法能够解决问题的范畴。通过最近的一些研究,我们描述了一些淡水系统微生物生态学的示例,并强调分子手段如何提供新的生态学见解。最后,我们详细介绍了该研究领域的一些有前景的发展,以及这些发展如何影响淡水微生物生态学的未来研究。 7 B' n; V7 [/ j3 g$ Q. m [: o! }
原文信息- P& S9 f& u3 j' k; H
原名:Streams of data from drops of water: 21st century molecular microbial ecology
' m p. m3 ~$ j! I w* p9 ^ 期刊:Wiley Interdisciplinary Reviews-Water 【IF=3.943】
- | p) V* X! p$ A# J+ f) Y 通讯作者:Dave R. Clark 和Alex J. Dumbrell【英国埃塞克斯大学】
& }# I4 L9 p) m, ?6 D* D; e" e 时间:2018-02-24
( V5 Y! b# W8 m! f# T DOI: https://doi.org/10.1002/wat2.1280
! v9 |9 g1 \9 O; | I3 B. A 1.研究背景
( `* H+ \9 J* l9 j/ v$ F 1 | INTRODUCTION 0 E8 J% x) q# _2 `
微生物无处不在,是地球上最多样化的生物。据估计,全球有超过1030个微生物,并且有些微生物占据最初被认为没有生命的栖息地。可以说,微生物在生态学上是最重要的生物,已知它们支持一系列生态过程和功能,同时驱动主要的生物地球化学循环,因而对生态系统产生巨大影响。 0 ]6 X# I {7 {7 O
淡水生态系统对人类文明的持久性至关重要,但对环境变化最为敏感。此外,作为陆地和海洋环境之间的管道,上述任何一个生物群落中的环境变化都可能显着改变淡水生态系统。全球人口密度的增加和相关的环境变化将对淡水生态系统产生潜在的影响。微生物对于生态学家来说具有挑战性,因为他们的研究物种由于体积小,缺乏显著的形态特征而难以识别。为了应对这些挑战,已经开发了基于研究大分子的多种分子方法,并且现在常规应用于淡水生态系统中的微生物生态学研究。
- U, X: d( ` ~$ Z% \% i 最近的进展集中于开发越来越高分辨率和高通量的方法,为微生物生态学家提供了大量的分子技术工具。然而,对于不熟悉的分子技术的快速更新,以及分析这些数据所需的一系列伴随的生物信息学方法,可能令人困惑,需要进一步阐述分析加以理解。
% P: N' s* U0 P% C$ f 2.微生物生态学简史
2 f/ Z3 x8 I" g% }( X; ` 2 | A BRIEF HISTORY OF MICROBIAL ECOLOGY
" i$ m* |" ~4 m 用于研究微生物多样性的技术可分为两大类:依赖培养(culture-dependent)和与不依赖培养(culture-independent)。17世纪中期显微镜的发明促进了对先前未见过的微生物世界的首次观察。此后依赖培养方法迅速发展,重点是从环境中分离微生物,在显微镜下观察它们并进行生化实验。这些方法提供有关生长速率,代谢途径,营养需求和最佳生长条件的有价值信息,这些信息不可能从不依赖培养的方法中获得。因此,培养基培养仍然是微生物学的重要工具,当与不依赖培养的分子方法结合使用时,可以揭示以前未知微生物的生态学。然而,作为一种独立的方法,培养依赖的应用是有限的。通常不可能在实验室中重建复杂的自然环境,并且许多微生物具有高度特异的生长条件。因此,大多数微生物不易培养,因此,不依赖培养的分子方法是研究环境微生物群落所必需的。 & k6 K1 S i. s* G
不依赖培养的技术不需要从收集的环境样品中分离和培养微生物。这些技术中的大多数始于分子生物标志物的提取,来自所有生物体的基因,基因组,转录物和转录组的信息存在于其中。现在可以将分子技术应用于这些样品,其选择主要取决于所研究的问题以及可获得的时间和资源。通常,这些方法针对DNA,允许研究人员研究样本的分类学和功能多样性,无论DNA是来自死亡,活着,代谢活跃还是休眠的生物。或者,可以使用RNA,更清晰地了解微生物群落的代谢活性成分的分类学和功能多样性。
! E: m% d; ~1 `( B+ |2 y. [ 样品处理的下一步涉及通过聚合酶链式反应(PCR)扩增提取的核酸,PCR使得目标基因组/转录组区域的数百万拷贝扩增,以进行后续分析。扩增子目标的选择完全取决于所研究的问题,这将集中于提供有关生物进化特性或其功能潜力信息的遗传标记。此外,通过适当的引物设计,可以针对特定的微生物类群,使PCR方法成为筛选水样中病原微生物存在的有效方法。通过实时定量PCR(qPCR)的技术,可以实现基因拷贝数在扩增时的定量,从而提供额外的和互补的分类或功能组成信息。
+ { [" o u1 V- Y7 a2 ?7 F4 R) z 在过去的20年中,测序技术和计算能力的快速发展已经改变了分子生态学领域。虽然使用克隆文库与第一代测序技术相结合只允许少量扩增子以连续方式测序,但下一代测序(NGS)技术现在允许数百万个扩增子并行测序。宏基因组学可以用于生物多样性调查(图1)。通过针对特定微生物类群,宏基因组方法在筛选和鉴定特定微生物方面也是有效的,在淡水生态系统的背景下,这对于监测潜在的污染源尤为重要,例如,通过检测污水中粪便来源的大肠菌群,或检测环境微生物群落中的抗生素抗性基因。此外,当应用于环境DNA时,可以使用宏基因组来快速确定可能无法观察到微生物的存在。 / b6 [" D2 b( o# p" b# o
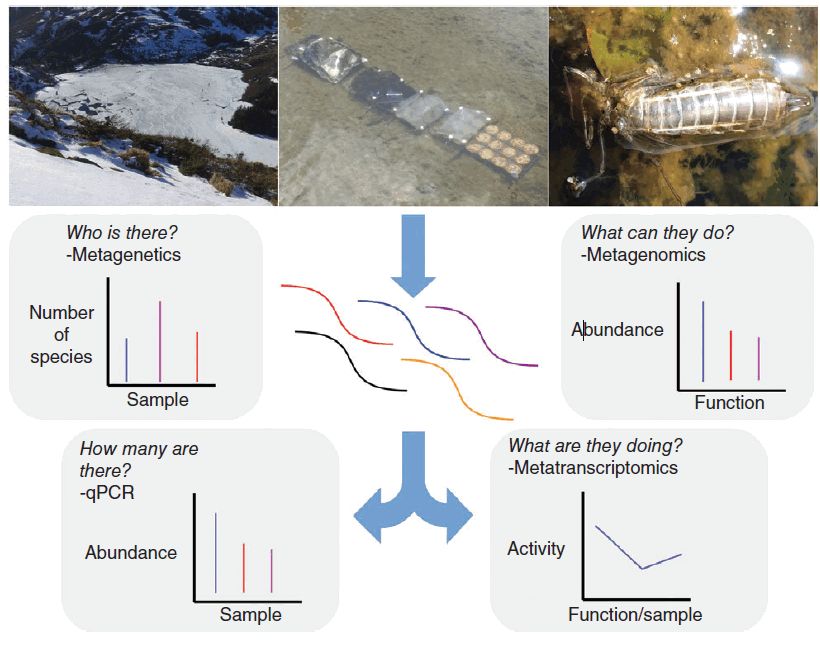
9 R: }: U, }, B. O" V
图1. 一些最常用的分子和宏基因组学方法示意图,以及它们适合解决的科学问题。
9 p2 S% [: C+ w9 J 无论该研究是观察性的,实验性的还是常规的生物监测,所有方法都需要从环境中提取和纯化生物分子,例如DNA或RNA。 左边的方法需要通过PCR扩增特定基因,而右边的方法不需要靶标,在整个核酸库水平上进行研究。
9 u0 Q' w0 k/ g) ~6 {8 w+ z NGS技术的出现也使人们能够轻松获得不仅PCR扩增子测序的单独方法,称为宏基因组学(metagenomics)。它将包含来自所有物种的基因组DNA,打断成较小的片段并直接测序(图1)。通过生物信息学组装得到的序列片段,可以获得较长的序列,如果获得足够的序列,甚至可以重建整个基因组。该方法的优点是更准确地鉴定存在的微生物,以及预测完整功能的能力。通过检查测序基因组的功能和代谢属性,群落的概况。另外,宏基因组方法避免了与PCR扩增相关的已知偏差,包括引物偏好或嵌合体形成。因此,从宏基因组序列数据生物信息学提取系统发育或功能标记基因可提供比遗传方法更少偏向于调查微生物多样性的方法。因此,宏基因组学作为研究微生物群落生态学的许多方面拥有巨大前景。
. V# V* o) l3 a6 [0 `7 A 如前所述,通过检测RNA而不是DNA,可以采用相同的(宏基因组)方法来量化在给定时间哪些基因被主动表达(图1)。因此,宏转录组学可以在给定时刻提供群落的功能和代谢活动的详细表达谱,并且可以用于检查响应于环境扰动或现有梯度的瞬时变化。在微妙的对比中,宏基因组学量化了群落的功能和代谢潜力,在更长的时间尺度上整合数据。除了这两种方法之外,通过研究群落内存在的蛋白质(宏蛋白质组学)和代谢物(宏代谢组学),替代“组学”方法可以提供对下一级生物组织的见解。 0 t4 V! Y3 u, ?) ~9 D2 s: q p
在过去的十年中,从单一NGS运行中获得的序列数量增加了几个数量级。从历史上看,罗氏454焦磷酸测序平台是分子微生物生态学中使用最广泛的高通量测序技术,但在2016年已停产。 Illumina的MiSeq和HiSeq平台现在在很大程度上占据了主导地位,在序列数量和运行成本方面与其他平台相比毫不逊色。随着每次运行数千至数百万甚至数十亿的序列数据集的增长,分析分子数据集的计算成本也大大增加。现代序列数据集的庞大规模意味着生物信息学分析现在最好在高性能计算机上进行,其中大量的硬盘空间和随机存取存储器(RAM)使研究人员能够有效地处理他们的数据。此外,通过利用现代计算机处理器中可用的多个计算核心,当前的生物信息学工具能够通过在核心之间“并行化”某些任务来显着加速分析。然而,当通过高通量平台进行分子分析时,充足的计算资源的可用性仍应是一个关键考虑因素。这些平台,加上高性能计算和生物信息学方法的不断发展,意味着分子生态学家现在有潜力探索微生物群落的结构和详细功能。 + N' D B {! ^- Y, u/ {8 U
3 .实例探究
* O; A1 F/ H9 ]4 @1 y 3 | CASE STUDIES & \6 Q1 l# F6 R4 M
3.1 | qPCR-肯尼河流域的灾难性农药泄漏事故
. d! C' S8 z6 i; Z 3.1 | qPCR—A catastrophic pesticide spill in the river Kennet
, Y2 `, k3 ~8 S' [, V4 s$ X 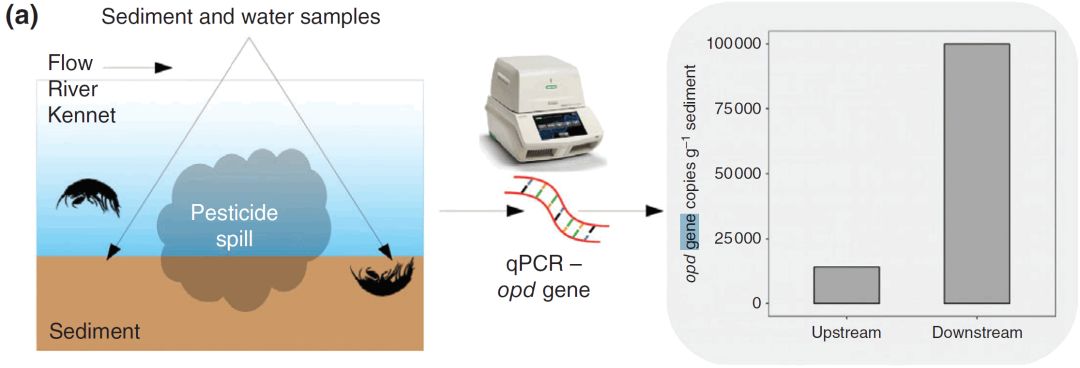
# b$ f/ I' O% t3 B7 h: v+ c4 p& i 图2. 这里介绍的每个案例研究的图形摘要。(a)Thompson等人(2016)使用opd基因的qPCR分析表明相对于对照沉积物,污染沉积物中农药降解微生物的丰度增加了7倍。 % D$ w. ~6 v L' d3 Q: a
在水生系统中,微生物构成食物网的基础,并且通常在环境扰动中表现出快速变化。因此,了解微生物群落如何应对环境变化,可以阐明生态影响如何通过食物网传播,同时提供有关微生物群落本身功能变化的信息。例如,, Thompson等人(2016)证明了应对肯尼特河(英国)内突然和灾难性的农药泄漏而导致的微生态变化。2013年夏天,在Kennet河内发现了杀虫剂的灾难性泄漏,摧毁了15公里长的河流中的所有无脊椎动物的生命。通过采集微生物群落,以及无脊椎动物和其他大型动物,Thompson等人试图从生物组织的各个层面(从基因到整个生态系统)了解这种泄漏的潜在影响。 qPCR对污染事件上游和下游微生物群落的分析表明,农药对微生物有直接和间接的影响。通过量化有机磷水解酶基因丰度的变化(opd;参与杀虫剂的降解)(图2a),Thompson等人表明,在泄漏后,能够降解农药的细菌增加了7倍。氨单氧化酶基因(amoA,参与氨的氧化)的定量揭示了氨氧化微生物的丰度(约30倍)的甚至更大的变化。作者将此归因于腐烂的无脊椎动物的质量增加,这将导致氨浓度增加。因此,通过靶向这两个微生物功能群,作者能够检验杀虫剂对微生物群落的直接和间接影响;并为观察到的生态系统功能变化提供解释[1]。
7 H; I& j D, w; y9 s 3.2 | 扩增子-厌氧氨氧化是淡水中重要的氮素流失途径
/ z" Z) ~9 B3 Q1 ` 3.2 | Metagenetics—Anammox as an important nitrogen loss pathway in freshwaters
% v( A8 e5 K) l( R# j% ~ 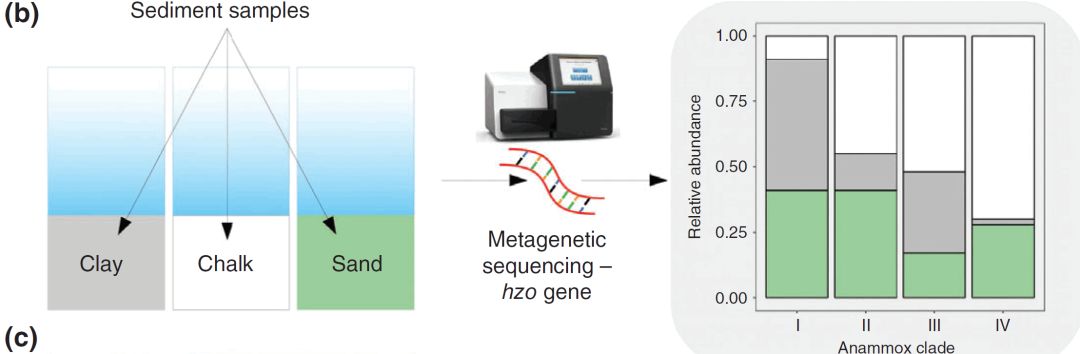
3 O8 Z- |! y: p4 U8 M% X (b)Lansdown等人(2016)对来自不同基础地质的河床沉积物的厌氧氨氧化(hzo)基因进行了测序。他们发现了四种不同的厌氧氨氧化细菌的系统发育分支,它们在不同的河床地质中的相对丰度明显不同。
: Q- c7 W% n/ Y l0 Y( |5 d# V NGS方法也有助于揭示淡水微生物群落中各种功能和代谢途径的存在。例如,Lansdown等人(2016)研究了氮气产生的替代途径 - 厌氧氨氧化(anammox) - 已知其在海洋环境中很重要,但在淡水中相对没有得到充分研究。 厌氧氨氧化是在没有氧气的情况下将氨转化为氮气而不进行反硝化和硝化的过程,这些过程代表了典型的氮循环。以前,该过程被认为发生在仅限于厌氧条件占主导地位的深海沉积物,直到最近才在淡水中观察到这一过程。
: ?, f7 F4 j+ e" _0 P Lansdown等人试图了解淡水流的基础地质是否影响厌氧氨氧化,以及厌氧氨氧化细菌的丰度和活动是如何支撑的。他们使用扩增子测序(图2b)研究了源自三种不同地质类型的沉积微生物 - 白垩,绿砂和粘土。在16S rRNA基因扩增子中未检测到已知的厌氧氨氧化细菌属,表明它们与其他细菌类群相比相对较少。然而,肼氧化还原酶基因(hzo; 厌氧氨氧化所需)的扩增子测序显示存在四种不同的厌氧氨氧化细菌进化分枝,其在不同的地质过程中相对丰度发生变化(图2b)。这些结果支持厌氧氨氧化过程速率的测量,并揭示厌氧氨氧化物是白垩地质中氮气损失的主要途径[2]。
8 |" I$ p4 R; E) J! k1 f 3.3 |宏转录组学-淡水微生物的功能多样性
. [! p* o! |, m* l+ t4 p2 N& \ 3.3 | Metatranscriptomics—Diel functional diversity of freshwater microbes
6 o [2 ~! K( i4 n; o* V 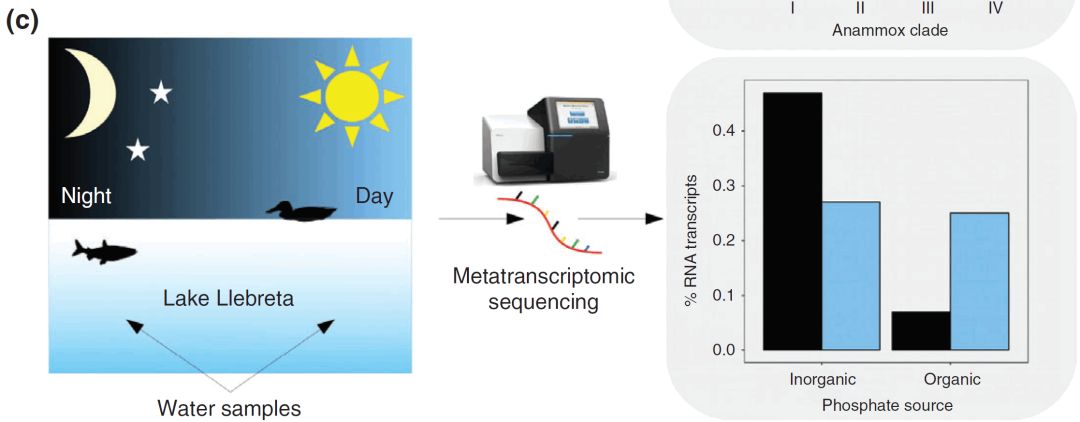
* k6 Q, Q; F: G0 C: Y (c)Vila-Costa(2013)在进行了湖泊浮游生物的宏转录组测序, 他们发现磷酸盐摄取基因根据磷酸盐来源和一天中的时间差异表达。 ; t1 g) u' e3 g5 i) \0 x
尽管宏基因组学具有很高的信息量,但通过宏转录组学方法可以更好地回答解决大规模微生物功能多样性模式的研究问题。例如,Vila-Costa等人(2013年)通过对Llebreta湖的转录组学分析,提供了对生态策略中吸收和代谢必需营养磷(P)以及其他关键功能的综合评估。该淡水湖位于西班牙。他们检测到白天相对于夜间光合异养过程的基因表达增加,以及与无机磷摄取相关的更高基因表达(图2c)。在夜间相反,转变为表达增加参与有机磷摄取和代谢的基因(图2c),从而表明在24小时内细菌P代谢的不同生态策略。此外,无论采样时间如何,属于细菌类的拟杆菌纲(Bacteroidetes)和β-变形菌纲(Betaproteobacteria)的转录本都是最丰富的,提供了对活跃的淡水细菌群落的概况的描述[3]。目前,宏转录组学的局限性可能是特定蛋白质的低注释和描述,特别是在淡水微生物群落中。这主要是由于淡水参考数据库的稀缺,需要大量研究人员进行研究,但随着该领域的不断发展,无疑将会做的更好。
6 l' Q+ U: C/ n$ v5 z7 Y+ N: \ 4.分子微生物生态学的未来- U& {# T7 ^' x- R0 m
4 | THE FUTURE OF MOLECULAR MICROBIAL ECOLOGY
) U5 w! m- R9 g( C4 J9 E# ? 在过去二十年中,DNA / RNA测序技术的发展异常迅速。例如,如前所述,罗氏的454焦磷酸测序仪 - 一种用于许多早期NGS研究的高通量测序平台 - 仅在使用十年后就停产了;被更便宜、更新的NGS平台取代。在这里,我们重点介绍一些可能在未来几年推动该领域发展的最有希望的发展技术。分子工具很可能为生物监测方法的“下一代”奠定基础,可能彻底改变淡水生态系统的常规监测和管理[4,5]。这些方法大量借鉴了最初开发用于量化微生物多样性的方法,但扩展到同时检测从微观到宏观生物的所有分类群。这种方法从环境样品中分离的核酸包含源自该生态系统中存在的所有生物的粪便,粘液,脱落组织或腐烂物质等环境DNA(eDNA)。因此,对PCR扩增的微小修改可以定量样品中存在的所有物种。这些方法的潜力巨大,克服与分类学专业知识的可用性相关的问题,以及当前生物监测方法的非标准化。因此,使用eDNA方法量化生物多样性正在迅速增长并推动持续的方法改进,并将很快成为淡水栖息地的常规方法。 & C8 E- ?4 r: s0 O; v3 ?9 \$ U
虽然分子工具在监测淡水生物多样性方面有巨大潜力,但只检查食物网中的微生物成分可以快速、实时地衡量当代生态系统的健康状况。如前所述(参见案例研究),微生物群落几乎立即对环境变化做出反应,从而为环境管理部门提供了一个“警告”系统。实际上,许多化学应激物进入食物网的途径是通过基础微生物的生物膜。这些不是新概念,微生物真核生物,如硅藻,通常用于生物监测。例如,营养硅藻指数(TDI)表示水体的营养状态,并且基于与不同营养状态的水相关的某些硅藻类群的存在。然而,TDI需要耗时的形态识别一组具有挑战性的生物,并且通过分子方法更新因形态学和分子分类学之间的脱节而受到阻碍[6,7]。然而,随着技术的发展,我们可预见到克服目前的限制,并通过对微生物群落的常规监测,可快速检测淡水生态系统中的新应激源。 }5 [2 \! ~( {: h
通过提供便携式现场技术,未来的发展也可以减少对专用分子实验室的需求[4]。例如,Nanopore MinION已经为研究人员提供了现场NGS测序能力,但需要改进序列质量[8]。然而,PCR技术的小型化目前允许对于宏基因组学和qPCR进行现场核酸扩增[9]。这些开发与易于编程的微型计算机(例如,Raspberry Pi)[10]相结合,使全自动,远程分子生物监测的有趣前景更接近现实。这有可能更快地识别环境扰动,从而更快地实施补救措施,同时减少可能影响微生物群落组成的样品处理和储存问题。后一点对于基于RNA的方法(例如,宏转录组学)特别重要,因为其在专用-80℃储存之外的半衰期短且不稳定。 . R5 z* J6 O7 F" h8 x6 Z
直到最近,由于缺乏分类学注释的功能基因数据库,连接系统发育和功能基因数据一直具有挑战性,促使生物信息学方法试图“预测”来自系统发育序列数据的功能性状,例如PICRUSt。随后,将功能基因与系统发育信息基因全面连接的唯一方法是分离筛选感兴趣的生物体,并对其整个基因组进行测序,这受到大多数微生物(缺乏)可培养性的限制。然而,epicPCR可以通过在分离核酸之前捕获乳液液滴中的单个微小细胞来克服这种限制,从而允许系统发育和功能基因的共扩增[11]。或者,单细胞基因组学方法能够在基因组测序之前从混合群体中分离单个微生物细胞(例如,通过流式细胞术),从而消除了在纯培养物中分离细胞的需要。单细胞“组学”方法已经揭示了淡水系统中生态重要微生物的未知功能[12]。高通量与单细胞“组学”的耦合引入了对水生微生物群落功能和系统发育的研究拥有诱人的前景[13]。这些方法提供了同时评估微生物分类学和功能多样性的新颖且潜在的高通量方法;可以说是实现了具有生态意义的数据的黄金标准。
4 ^" E% x' h \$ M* u, U 5.结论% O- H4 k: P; ?$ r/ q3 C2 v
5 | CONCLUDING REMARKS 7 z% J/ x/ Z5 G& W! K
分子技术的迅速发展提供前所未有的详细研究淡水微生物群落的工具。随着微生物在其中发挥的关键作用,从而对淡水栖息地进行更全面的了解。目前,分子微生物生态学的发展正在更广泛的生态研究界得到应用;如改进生物监测,重新定义食物网,并检查以前忽略的微生物类群的生态网络。作用于淡水生态系统的多种压力源的持续增加导致环境管理面临越来越大的挑战,但随着我们可以使用的最新分子工具的产生,微生物群落在满足和理解这些挑战中的作用将永远不会被忽视。
# D/ I! U$ H H. g2 o 参考文献. F1 Y4 t! j, l6 ^+ T [
Thompson, M. S., Bankier, C., Bell, T., Dumbrell, A. J., et al. (2016). Gene-to-ecosystem impacts of a catastrophic pesticide spill:Testing a multilevel bioassessment approach in a river ecosystem. [J]Freshwater Biology, 61(12), 2037–2050
! z5 P5 t' u- N Lansdown, K., McKew, B. A., Whitby, C., et al. (2016). Importance and controls of anaerobic ammonium oxidation influenced by riverbed geology. [J] Nature Geoscience, 9(5), 357–360
9 o- r7 |+ _; Z2 B4 ~5 ~ Vila-Costa, M., Sharma, S., Moran, M. A., et al. (2013). Diel gene expression profiles of a phosphorus limited mountain lake using metatranscriptomics. [J]Environmental Microbiology, 15(4), 1190–1203. 2 q8 B* z# h' n5 k/ X" A9 `
Bohan, D. A., Vacher, C., Tamaddoni-Nezhad, A., et al. (2017). Next-generation global biomonitoring: Large-scale, automated reconstruction of ecological networks. [J]Trends in Ecology & Evolution, 32(7), 477–487. * p2 [2 `0 h: R. f
Jackson, M. C., Weyl, O. L. F., Altermatt, F., et al. (2016). Chapter twelve-recommendations for the next generation of global freshwater biological monitoring tools. [J]Advances in Ecological Research, 55, 615–636.
; U' j- ~7 b3 S2 q0 Q) F Apothéloz-Perret-Gentil, L., Cordonier, A., Straub, F., Iseli, J., et al. (2017). Taxonomy-free molecular diatom index for high-throughput eDNA biomonitoring. [J]Molecular Ecology Resources, 17(6), 1231–1242.
; P0 z& v8 P8 W Visco, J. A., Apothéloz-Perret-Gentil, L., Cordonier, A., et al. (2015). Environmental monitoring: Inferring the diatom index from next-generation sequencing data.[J] Environmental Science & Technology, 49(13), 7597–7605. 6 M3 [6 m/ N7 g6 u0 ~$ Y1 w& u
Mikheyev, A. S., & Tin, M. M. (2014). A first look at the Oxford Nanopore MinION sequencer. .[J] Molecular Ecology Resources, 14(6), 1097–1102. 8 y3 }; z' C% S. u
Marx, V. (2015). PCR heads into the field. [J] Nature Methods, 12(5), 393–397.
% x" `: A. ^2 O/ o- C$ }# j Cressey, D. (2017). The DIY electronics transforming research. [J] Nature News, 544(7648), 125–126 , C) A, p" T8 \6 z& [
Spencer, S. J., Tamminen, M. V., Preheim, S. P., et al. (2016). Massively parallel sequencing of single cells by epicPCR links functional genes with phylogenetic markers. [J] The ISME Journal, 10(2), 427–436
{1 Q- h5 F. M8 k/ H Ghylin, T. W., Garcia, S. L., Moya, F., et al. (2014). Comparative single-cell genomics reveals potential ecological niches for the freshwater acI Actinobacteria lineage.[J] The ISME Journal, 8(12), 2503–2516.
9 M/ ?' B- y: O% O Lan, F., Demaree, B., Ahmed, N., & Abate, A. R. (2017). Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. [J]Nature Biotechnology, 35, 640–646.
- K% i' k9 w3 }6 ~! o1 Y 猜你喜欢& ~4 u. H2 [0 L) m6 D* a
10000+:菌群分析宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑 9 @( o+ A/ [4 M# f
系列教程:微生物组入门 Biostar 微生物组 宏基因组 % _$ ]/ e" O7 O+ w: m( V9 M$ s
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
# C2 \# z! Z/ Q$ z8 }2 G2 \ 一文读懂:宏基因组 寄生虫益处 进化树 " {# z2 A s; A! c0 g6 }5 Y7 T
必备技能:提问 搜索 Endnote 7 R7 h3 Y- X* S+ r1 y/ H
文献阅读 热心肠 SemanticScholar Geenmedical 7 Q3 p$ x& ~$ ?1 h
扩增子分析:图表解读 分析流程 统计绘图 ; f* R; Q' M) I$ h# W. u
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
" t! t" T, C: J! z 在线工具:16S预测培养基 生信绘图
0 k8 I" @) S7 v$ y1 {, `3 s# S 科研经验:云笔记 云协作 公众号 # z' F5 y1 ~5 F' o) E7 T) P! a! w
编程模板: Shell R Perl # I+ S, a* p/ M' ~+ \
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
+ ^& f" n# R! ?* Z! M4 B 写在后面
& v2 s: ^6 m- x* R8 a9 S% a 为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》
7 D1 f% a* B- a9 \ d1 \. V* k# l 学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。 # H7 _$ [3 S2 t3 S2 t* |' [
9 w* L" R; r5 b9 V' n) A& U

1 b( r h5 j9 j# {9 q: A8 \& \ 学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组” / g9 {" M6 V/ \
) A2 {, z. ^3 D1 n# _7 x7 S7 a5 ~
 点击阅读原文,跳转最新文章目录阅读 点击阅读原文,跳转最新文章目录阅读
4 M8 P1 U# d0 S% Y; w3 D1 m
* ?' ^ T# Z4 O# t: `) H. ~0 S
1 E1 E; e6 t" h3 n \/ h | 

