|
; g# h* D" q% Y3 _" v     
' D4 H+ Y3 z* \! W 点击关注:复层矿脂包覆防腐蚀技术(PTC) ; I( O2 f+ c6 n, k8 U( M3 O5 c
点击报名:2022第九届海洋材料与腐蚀防护大会暨2022第三届钢筋混凝土耐久性与设施服役安全大会
, ~4 t6 N/ c+ p7 g: O, b1 A7 Q3 E 作者:许竞翔,冯建方,孙强,褚振华 . ]! j, I/ m( ^' S8 G6 M" }
上海海洋大学工程学院
" O4 W; f5 _6 g0 U/ J 0 前 言 海洋环境十分恶劣,金属表面水含量、温度、辐照度、氯离子浓度、SO2等污染物等环境因素对金属材料的腐蚀有很强的影响,从而对其力学性能和几何形状等造成不同程度的影响[1,2],造成设施结构损伤,使役寿命缩短,严重时危害生产安全,并造成相当大的财政负担[3],其中船舶和海洋工程装备中金属材料的腐蚀防护成本占比很高,对其腐蚀机理的研究尤显重要。长期以来,有关金属材料腐蚀的研究大多数是通过试件实验进行的,主要是通过模拟实际环境条件,观察金属材料的特征和变化,从腐蚀产物、腐蚀表面状态、腐蚀产物的演变等方面来研究各种金属材料在不同环境中的腐蚀机制和原理[4,5,6,7,8]。但是金属发生腐蚀的环境十分复杂,受到众多因素以及因素之间耦合作用的影响,任何单一因素对腐蚀行为的影响都可能受其他因素的干涉,腐蚀的机理不太清晰,且腐蚀实验所需成本往往较高。数值计算模拟相对成本较低,该方法从原子/分子尺度研究腐蚀在微观尺度上的动态变化过程,如金属与环境的化学反应、化学键的断裂和形成、表面的缺陷和扩散、应力应变的变化等等。随着计算机技术的快速发展,数值模拟已经成为研究腐蚀等领域的方法之一,其中分子动力学和密度泛函(DFT)理论等是主要的数值模拟方法[9,10,11,12,13]。金属的腐蚀是指金属和环境之间发生化学或电化学反应,金属成为离子态或与其他粒子结合形成化合物,其实质是原子间化学键的断裂和形成,现有的研究[9,10,11,12,13]充分证明分子动力学有能力描述金属的腐蚀过程。分子动力学模拟是通过构造出比较理想的模型,改变单一变量(如温度、表面取向、氯离子浓度等)模拟在真实环境中金属材料腐蚀所发生的动态过程,然后与实验研究对比,明确腐蚀机理,从而找到更加高效的抑制或减缓腐蚀发生的方法[13]。分子动力学模拟已成为研究金属材料腐蚀的重要方法,目前已成功用于摩擦磨损[14]、超精密加工[15]、微纳米元器件[16]等研究领域。
8 s3 [. i( L+ u 1 原子间的势模型 分子动力学算法的基础,在于从原子间相互作用势函数出发,基于牛顿第二定律来求解体系中各原子的运动轨迹,进而获取体系随时间的演化过程。原子间相互作用势函数的可靠性直接决定所得原子运动轨迹的准确性,从而在分子动力学模拟中具有非常核心的作用[17,18,19,20]。在多粒子系统中,牛顿第二运动定律的二阶微分方程定义为[21,22]:Fi=miai (1)式中:Fi是原子i的力,mi和ai分别是原子i的质量和加速度。用有限差分法对式(1)中的各自由度进行时间积分:Fi=-∇iV (2)式中:V是原子间势能函数,Fi是其梯度。原子间势能函数的计算是分子动力学模拟中计算量最大的一步,通常占整个模拟时间的95%[21]。描述原子间相互作用的势函数很多,早期主要是通过两体势函数[23]描述,如Lennard-Tones(LJ)势和Morse势等。但是两体势函数在描述原子间化学键的断裂和形成方面还有些不足。随着研究和发展,出现了考虑角度效应和静电势等的多体势函数,而描述化学反应较好的势函数主要有修正的嵌入原子法[23,24](Modified Embedded Atom Method, MEAM)和反应力场(Reaction Force Field, ReaxFF)势函数等等[25]。 1.1 MEAM势函数嵌入原子法(Embedded Atom Method, EAM)势函数主要描述的是所有原子核之间的排斥作用和原子的原子核与核外电子及原子周边其他电子的静电作用,具体形式通过拟合完整纯金属体系的点阵常数、升华能、弹性常数、空位形成能、合金潜热等基本物理量来定义[23]。EAM势函数定义为[24]:EEAΜ=∑iE(ρi)+12∑j>iϕ(rij) (3)式中:EEAM表示系统的总能量,i和j分别表示两原子,E表示将原子i嵌入到电子密度为ρi的位置处i所需要的嵌入能,ϕ表示相距r的i和j原子之间的两体势。而在修正的嵌入原子法(MEAM)中,系统的总能量为嵌入原子势和静电势(Electrostatic Potential, ES)之和。MEAM势函数定义为[24]:Esystem=EEAM+EES (4)式中:Esystem表示修正后的系统的总能量,EEAM和EES分别表示嵌入原子势能和静电势能。EES定义为[24]:EES=∑iqi{χi0+∑j≠iΖj[ωi(rij)-μij(rij)]}+12∑iqi2Ji0+12∑j≠iqiqj[1rij+μij(rij)] (5)式中:Zj表示有效的原子核电荷,一阶导数χ是电负性,二阶导数J与自库仑斥力相关,ωi (rij)表示短距离核吸引力积分,μij(rij)表示短距离库仑相互作用积分,qi是离子i的电荷。Lee等[26]通过MEAM开发了Fe-Al二元体系的原子间势,该电势可以描述Fe-Al二元合金的各种基本物理特性-结构、弹性和热力学特性、缺陷形成行为以及缺陷之间的相互作用,且这些性能与实验数据和第一性原理计算的数据基本吻合,证明了该方法从原子角度研究各种缺陷形成行为及其对高铝钢以及Fe-Al二元合金的力学性能的影响方面的适用性。Jelinek等[27]采用MEAM开发了铝、硅、镁、铜和铁的相互作用势,根据从头算(ab initio method, 基于量子力学基本原理直接求解薛定谔方程的量子化学计算方法)得到的NaCl参考结构中元素对的结构和弹性特性,建立了元素对的MEAM参数,并进行了调整,重现了最稳定二元化合物的从头计算生成热。然后对几种二元化合物的缺陷形成能、平衡体积、弹性模量和生成热与从头算模拟和实验结果比较,验证了MEAM势的可行性。Campbell等[24]通过MEAM模拟半径为10 nm的铝纳米团簇的氧化过程,基于电负性均衡原理考虑了铝和氧之间电荷转移的影响。铝向表面扩散,而氧向团簇内部扩散,铝的扩散率比氧在氧化物中的扩散率高30%~60%。氧化渗滤的过程分为3步,分别为从氧气分子吸附到铝表面、氧原子通过fcc铝的四面体和八面体的位置向基体扩散、最后形成[OAl4]四面体簇,氧化膜厚度随时间的变化如图1所示,该氧化过程解释了在典型模拟的早期阶段氧化物生长的减速,同时也解释了为何氧化层的厚度和平均温度随时间的延长而不断增加。 1.1 MEAM势函数嵌入原子法(Embedded Atom Method, EAM)势函数主要描述的是所有原子核之间的排斥作用和原子的原子核与核外电子及原子周边其他电子的静电作用,具体形式通过拟合完整纯金属体系的点阵常数、升华能、弹性常数、空位形成能、合金潜热等基本物理量来定义[23]。EAM势函数定义为[24]:EEAΜ=∑iE(ρi)+12∑j>iϕ(rij) (3)式中:EEAM表示系统的总能量,i和j分别表示两原子,E表示将原子i嵌入到电子密度为ρi的位置处i所需要的嵌入能,ϕ表示相距r的i和j原子之间的两体势。而在修正的嵌入原子法(MEAM)中,系统的总能量为嵌入原子势和静电势(Electrostatic Potential, ES)之和。MEAM势函数定义为[24]:Esystem=EEAM+EES (4)式中:Esystem表示修正后的系统的总能量,EEAM和EES分别表示嵌入原子势能和静电势能。EES定义为[24]:EES=∑iqi{χi0+∑j≠iΖj[ωi(rij)-μij(rij)]}+12∑iqi2Ji0+12∑j≠iqiqj[1rij+μij(rij)] (5)式中:Zj表示有效的原子核电荷,一阶导数χ是电负性,二阶导数J与自库仑斥力相关,ωi (rij)表示短距离核吸引力积分,μij(rij)表示短距离库仑相互作用积分,qi是离子i的电荷。Lee等[26]通过MEAM开发了Fe-Al二元体系的原子间势,该电势可以描述Fe-Al二元合金的各种基本物理特性-结构、弹性和热力学特性、缺陷形成行为以及缺陷之间的相互作用,且这些性能与实验数据和第一性原理计算的数据基本吻合,证明了该方法从原子角度研究各种缺陷形成行为及其对高铝钢以及Fe-Al二元合金的力学性能的影响方面的适用性。Jelinek等[27]采用MEAM开发了铝、硅、镁、铜和铁的相互作用势,根据从头算(ab initio method, 基于量子力学基本原理直接求解薛定谔方程的量子化学计算方法)得到的NaCl参考结构中元素对的结构和弹性特性,建立了元素对的MEAM参数,并进行了调整,重现了最稳定二元化合物的从头计算生成热。然后对几种二元化合物的缺陷形成能、平衡体积、弹性模量和生成热与从头算模拟和实验结果比较,验证了MEAM势的可行性。Campbell等[24]通过MEAM模拟半径为10 nm的铝纳米团簇的氧化过程,基于电负性均衡原理考虑了铝和氧之间电荷转移的影响。铝向表面扩散,而氧向团簇内部扩散,铝的扩散率比氧在氧化物中的扩散率高30%~60%。氧化渗滤的过程分为3步,分别为从氧气分子吸附到铝表面、氧原子通过fcc铝的四面体和八面体的位置向基体扩散、最后形成[OAl4]四面体簇,氧化膜厚度随时间的变化如图1所示,该氧化过程解释了在典型模拟的早期阶段氧化物生长的减速,同时也解释了为何氧化层的厚度和平均温度随时间的延长而不断增加。 $ ]6 D! R: v3 q2 Z
$ ]6 D! R: v3 q2 Z 图1 氧化膜厚度随时间的变化[24]
) ~3 P6 f; I# S# X# E7 Q, x1 J Fig. 1 Variation of oxide film thickness with time[24] 1.2 ReaxFF势函数反应力场(ReaxFF)是专门设计出来描述化学反应性、化学键的断裂和形成、表面、缺陷、扩散等的一种特殊类型的力场[25],它同时考虑了非键合和键合的能量函数。它通过能量描述化学键,有着较高的计算效率,弥补了量子化学方法和传统分子动力学之间的差距[25]。ReaxFF原子间势能函数定义为[25,28,29]:Esystem=Ebond+Eover+Eunder+Eval+Etor+Elp+EH-bond+Evdwaals+Ecoulomb (6)式中:Esystem表示系统的总能量,Ebond、Eover、Eunder、Eval、Etor、Elp、EH-bond、Evdwaals和Ecoulomb等能量项分别对应键能、欠配位的能量矫正项、过配位的能量矫正项、价角能、扭转角能、孤对能、氢键能、范德华能和库仑能。最后两个能量项范德华能Evdwaals和库仑能Ecoulomb计算所有原子对,而其他所有能项均由计算粒子i和j之间的键级获得。原子电荷是使用与几何有关的电荷分布来计算的,该分布通过电荷平衡法确定[30,31,32]。单个原子电荷和静电能E(q)随时间动态地变化。相应的静电能E(q)定义为:E(q)=Νi=1[wiqi+uiq2i+Τap(rij)Κc] (7)式中:qi是离子i的电荷;N是离子的总数;q是电荷长度N的矢量;wi和ui分别是离子i的电负性和硬度。Tap(rij)是一个七阶锥度函数,当带电粒在非键合截止半径内或外移动时,避免能量不连续;Kc是介电常数;rij是原子i和j之间的距离。原子电荷与系统的几何位置有关,在每一时间步上都进行计算。Nabankur等[33]利用ReaxFF分子动力学模拟研究了阳离子Li+、N a+、K+和阴离子Cl -对水的结构和动力学特性的影响,且已经成功地描述了这些系统固有的化学反应和扩散行为,能够将氢键动力学的定性趋势与不同体系的物种分布联系起来,使得其他电解质-水系统和热力学状态的模拟成为可能。Gabriele等[34]通过ReaxFF描述了银-氧和银-硫反应机理,Ag-O和Ag-S的反应快照如图2所示。 % R( T1 f9 d' z% w5 y) k1 E
% R( T1 f9 d' z% w5 y) k1 E 图2 Ag-O和Ag-S的反应快照
% ]6 J; x2 I( O1 \) G4 L Fig. 2 Reaction snapshot of Ag-O and Ag-S 图2中下方基底处较大的原子代表银原子,图2a中中上方小原子代表硫原子,图2b中中上方小原子代表氧原子。由图2可知Ag-O和Ag-S具有不同的反应机理和反应速率。Ag2O的生长必须通过氧化物表面的O2解离和O通过氧化物层向氧化物/Ag间相扩散进行,O2的解离黏着系数随着氧覆盖率的增加而急剧下降。一旦S8分子接近银,二者就不可避免地结合并发生反应,而且在第一层硫化银形成之后,硫化银的形成也相对较快,这是因为硫化物在银表面以外的生长不需要缺陷的存在。这个模拟证明了银极易受到硫腐蚀(失去光泽),而很难与O2发生反应。) I1 V+ _6 s0 _+ W+ G1 g( X
 
4 u0 y5 \( ]+ G2 T, u 
, }, l! c( u( D( E% m0 `
2 金属腐蚀分子动力学模拟模型 建立合适的金属腐蚀分子动力学反应模型是得到正确和可靠的模拟结构的首要条件。目前用于金属腐蚀的分子动力学模拟的反应模型主要有3种,分别为金属团簇-溶液/气体反应模型、溶液/气体-金属表面-溶液/气体反应模型和金属表面-溶液/气体反应模型。2.1 金属团簇-溶液/气体反应模型图3为金属团簇-溶液/气体反应模型的截面示意图。在该模型中,溶液/气体中的原子与金属团簇表面直接接触反应。Li等[35]使用金属团簇-溶液/气体反应模型研究铝团簇与水分子的反应,采用分子动力学的方法模拟了不同数量的水分子与铝团簇的反应行为。 & _! V# X2 S2 R/ F0 u) I: J & _! V# X2 S2 R/ F0 u) I: J
图3 金属团簇-溶液/气体反应模型的截面示意图 ^; ^8 `# }4 t1 L) Y) }
Fig. 3 Schematic diagram of the middle section of the metal cluster-solution/gas reaction model 2.2 溶液/气体-金属表面-溶液/气体反应模型图4为溶液/气体-金属表面-溶液/气体反应模型的示意图。在该模型中,溶液/气体中的原子与金属不同表面取向的表面(双面)直接接触反应。Liu等[36]使用溶液/气体-金属表面-溶液/气体模型研究铝(100)与O2的反应,采用分子动力学的方法模拟了铝的氧化过程。
" r5 r/ e6 C) K8 s6 O( ?- @ 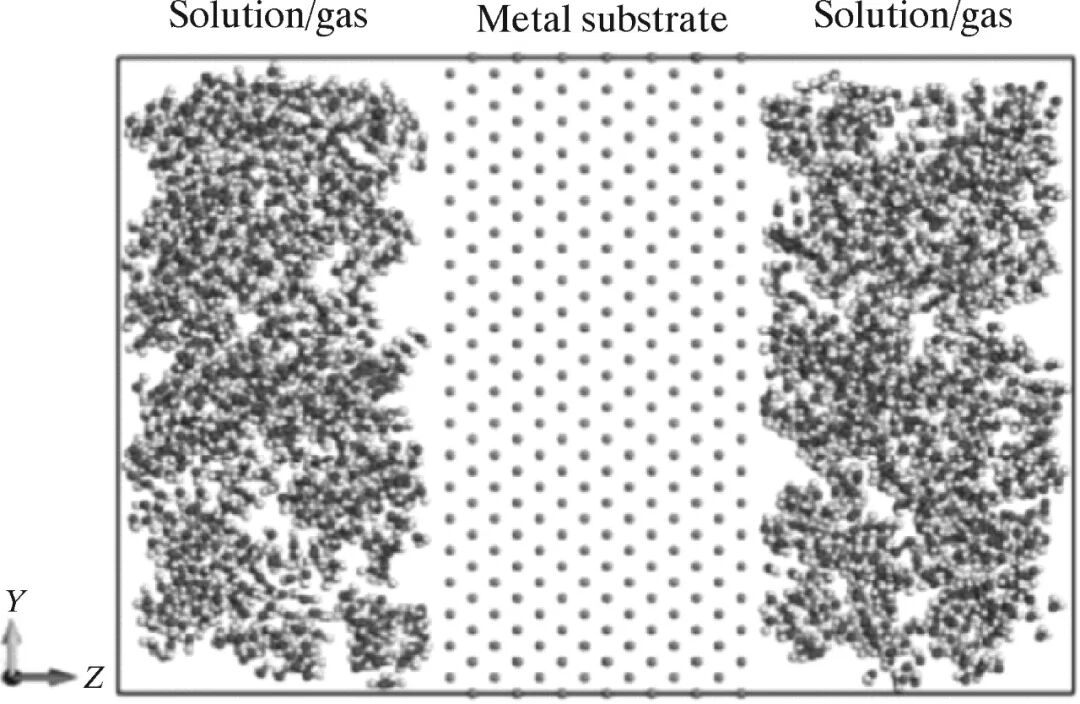
8 ^. U+ H0 D8 O ~5 K6 X) U 图4 溶液/气体-金属表面-溶液/气体反应模型的示意图
3 u* ~; R2 [& i7 a7 F8 m6 w( |# v Fig. 4 Schematic diagram of solution/gas metal surface-solution/gas reaction model 2.3 金属表面-溶液/气体反应模型图5为金属表面-溶液/气体反应模型的示意图。在该模型中,溶液/气体中的原子与金属不同表面取向的表面(单面)直接接触反应,如图5所示,在图5的左边固定一部分金属基体,在图5的右边一定距离处设置一个反射墙,在两端分别预留一定距离的真空。这种设置的目的是让Z方向展现为非周期性。DorMohammadi等[37]使用金属表面-溶液/气体反应模型研究了铁基体不同的表面取向的表面与纯水的反应,采用分子动力学的方法模拟了铁基体在纯水中腐蚀的初始阶段。 fill=%23FFFFFF%3E%3Crect x=249 y=126 width=1 height=1%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E) 3 ^0 X3 S% ]. k* t
3 ^0 X3 S% ]. k* t 图5 金属表面-溶液/气体反应模型的示意图
7 v9 G' g. p# ?1 e5 Y Fig. 5 Schematic diagram of metal surface- solution/gas reaction model 从以上分析可以看出,对于不同的金属材料体系,需要按要求采取不同的反应模型。一般来说,在研究金属材料腐蚀的过程中,大多数采用金属表面-溶液/气体反应模型[25,26,27,32,34],因为在实际中,金属表面往往只有一层与环境(溶液)直接接触,所以金属表面-溶液/气体反应模型被认为最能真实反映宏观尺度的金属材料腐蚀。但是目前如何桥接原子尺度与真实宏观尺度成为一个重要的问题[35],这也是未来金属腐蚀分子动力学研究的重点方向之一。
: J6 E) K' z+ ^% N# q3 e6 [
" `1 ^, `% a* t# h L. Y( s; ], }
3 金属腐蚀分子动力学模拟研究进展 海洋工程装备常用金属材料有钢、铁、钛合金、铜合金、铝合金、镁合金等[38]。海洋中的海水是一种具有一定流速的腐蚀性非常强的天然电解质溶液[38]。国内外研究表明[23,38,39,40],金属表面水含量、温度、氯离子浓度、表面取向、外电场等因素对海洋金属材料腐蚀都有着显著的影响,且这些因素之间有耦合作用,使得腐蚀变得复杂化。但是分子动力学模拟可以确定单一因素对金属材料腐蚀的影响。3.1 金属表面水含量一般将海洋腐蚀区域分为海洋大气区、浪花飞溅区、潮差区、海水全浸区和海底泥沙区[38],在这些区域中金属材料表面有一层相对较厚的水膜。当金属材料表面的相对湿度达到临界相对湿度时,腐蚀速度将大幅上升[40]。目前采用分子动力学模拟的方法对金属表面水含量对金属材料腐蚀的影响的报道已经有很多,但是尚没有形成统一的结论。一些分子动力学模拟研究表明,部分金属材料如铁、铝等在纯水中会被腐蚀。DorMohammadi等[37]分别建立了铁(100)、(110)、(111)与纯水的金属溶液模型,图6是铁(110)基体与纯水的初始构型,图6中左边整齐排列的是铁原子,右边是水分子团簇。) G4 ^/ i. D! D# T1 f
图6 铁(110)基体和水的初始构型[37]
) H! Y8 R1 o: R Fig. 6 Initial configuration of iron (110) matrix and water[37] 模拟结果表明铁基体在初始阶段会被纯水腐蚀,且铁(100)、(110)、(111)基体表面随着反应的进行将生成铁的不同价态的氧化物,并有铁原子溶解到纯水中。Li等[35]建立了铝纳米团簇与纯水的金属团簇-溶液/气体反应模型,模拟结果表明铝团簇与纯水发生反应,铝的表面会生成氧化铝并伴随着氢气生成,在一段时间后,铝表面氧化膜覆盖团簇表面,铝团簇不再与水反应。另一些分子动力学模拟研究表明,镍等金属材料不会被水腐蚀。Assowe等[25,29]建立了镍(111)与纯水的金属溶液模型,模拟结果表明,在没有外电场的情况下,镍(111)基体表面不和水反应,也没有镍原子溶解到水中。从以上分析可以看出,金属表面水含量会影响部分金属的腐蚀。一些本身不与水反应的金属,在加入了外电场等其他的影响因素后被腐蚀。3.2 温 度温度升高,原子吸收能量,相对而言更活跃,原子之间化学反应速率变快,因此温度对金属材料的腐蚀也会产生显著的作用。地球表面的太阳辐射能大部分被海洋所吸收,昼夜交替、洋流、大气环流等使得海水的温差很大。在海洋工程装备工作的时候,局部温度可能很高。而受限于实验设备,海洋工程材料在极端温度下的腐蚀缺乏系统研究。分子动力学模拟可以很稳定地控制系统中的温度变化,所以分子动力学方法能够很好地研究温度对金属材料腐蚀的影响。一些分子动力学模拟研究表明,温度越高,海洋金属材料腐蚀的速率就越快。DorMohammadi等[37]建立了Fe(110)与纯水的金属溶液模型(如图6所示)。研究温度变化范围为300~350 K,在25 MeV/cm外电场下铁和水侧氧原子数的比较,水侧的铁原子数随时间的变化如图7所示。
2 J3 y" y& @ d d9 G. o 图7 不同温度下原子总数随时间的变化[37]
) ^& G1 ^5 {3 c! A Fig. 7 Change of total number of atoms with time at different temperatures[37] 从图7中可以看出,随着温度的升高,铁侧的氧原子数略有增加,但水侧的铁原子数几乎相同。图8为不同温度下150 ps 时25 MeV/cm外电场界面的铁侧氧原子密度和水侧铁原子密度与界面Z的关系图,结合图7a可知,随着温度的升高,铁的氧化略有增加。综上可得,温度的升高加快了铁的腐蚀速率,但是并没有增加铁的腐蚀程度。4 c& R( c3 ?' L2 ]' @" r
图8 不同温度下铁水界面上原子密度比较[37]
% ~7 f! ^. d. t. j# e$ Y# @# L Fig. 8 Comparison of atomic density at the interface of molten iron at different temperatures[37] 3.3 氯离子浓度海洋中含有大量的盐类,通常把海水近似地看作3.0%(质量分数,下同)或者3.5%的NaCl溶液[39]。但是在实际海洋环境中,氯离子的浓度是有变化的,在海洋金属材料表面有一层水膜,水膜的蒸发和凝聚会导致材料表面盐分浓缩,改变氯离子的浓度[38]。一些分子动力学研究表明,金属的腐蚀和氯离子的浓度有关。Jeon等[40]建立了 Cu (111)基体在氯离子溶液中的腐蚀模型,图9为氯离子浓度为10 mol/L的水溶液条件下铜腐蚀模拟的初始模型,研究了水相条件下铜基体与氯的相互作用。300 K下腐蚀模拟弛豫500 ps时,不同氯离子浓度的水性介质和铜基底之间的界面的侧视图如图10所示,图10中下方排列规则的原子代表铜原子,上方紧密排列的有3个原子和2个原子的分子分别代表水分子和氯气分子。由图10可知,腐蚀模拟弛豫500 ps时,在0 mol/L和1 mol/L氯离子的模型中,铜离子没有扩散到溶液中,在10 mol/L氯离子的模型中,铜发生腐蚀且有铜离子扩散到溶液中。
/ P0 `# G) l( I& \! b/ V3 Y5 r! ?: R% P 图9 氯离子浓度为10 mol/L的水溶液条件下铜腐蚀模拟的初始模型[40] " u2 o1 T1 \ S* b' K; O
Fig. 9 Initial model of copper corrosion simulation in aqueous solution with chloride ion concentration of 10 mol/L[40]
. q8 d' t" s/ j2 P, b" `& k' c 图10 300 K下腐蚀模拟弛豫500 ps时,不同氯离子浓度的水性介质和铜基底之间的界面的侧视图[40] . q% n) h3 ^6 ~9 Z
Fig. 10 Side view of the interface between the aqueous medium with different chloride ion concentrations and the copper substrate when the corrosion simulation relaxation was 500 ps at 300 K[40] DorMohammadi等[41]建立了Fe(110)基体在pH=13.5的碱性溶液中的模型,结果表明在Fe(110)表面形成了一层由富含Fe2+的内氧化物层和富含Fe3+的外氧化物层组成的氧化膜;在碱性溶液中加入氯离子以后,Fe(110)表面的氧化膜结构被破坏,图11为氯化物和氢氧化物与氧化铁表面相互作用的不同阶段。图11中被方框、菱形、五边形圈住的小球分别表示铁原子、氯原子和OH-。如图11所示,铁原子以Fe(OH)Cl2和FeCl3的形式与钝化膜分离,溶解到溶液中;氯化物在去钝化过程中有2个主要功能:首先,引起薄膜/电解质界面附近电解质的局部酸化。其次,引起暴露铁原子的溶解。2种作用都需要电解质中存在足够数量的氯离子;在电解质中使用1 mol/L和2 mol/L NaCl进行的模拟显示没有局部酸化和铁的溶解,表明太低的氯化物浓度不能引发铁表面氧化膜的腐蚀。4 [ C; \, J% \9 V& C5 c
图11 氯化物和氢氧化物与氧化铁表面相互作用的不同阶段 ; J$ ]- Y8 W* ~) v
Fig. 11 Different stages of interaction between chloride and hydroxide and iron oxide surface 3.4 表面取向腐蚀现象的本质是金属材料和环境中其他物质之间的化学反应,即原子间的键的断裂和形成,因此基体的晶体结构、表面原子结构的取向,以及溶液与晶体之间的取向均会对腐蚀产生影响。分子动力学模拟方法在研究表面取向对金属腐蚀的影响方面有着巨大的优势,国内外许多学者对此开展了研究。一些分子动力学模拟研究表明,在相同条件下,金属晶体不同的表面取向对金属基体的特性有不同的影响。Jeon等[42]分别建立了铁基体不同表面(100)、(110)、(111)的氧化模型,图12为铁(100)、(110)、(111)在氧化1 ns时的侧视图,图12中下方整齐排列的为铁原子,杂乱排列的为氧原子。模拟结果表明,在300 K和900 K下,Fe(110)显示出最多量的氧化物生长,Fe(111)次之,Fe(100)最少量(如图12所示)。DorMohammadi等[37]分别建立了Fe(100)、(110)、(111)与纯水的金属溶液模型(如图6所示),模拟结果表明,不同的铁表面取向对于铁的氧化过程和电荷分布没有明显的影响。综合分析得到,铁基体的不同表面取向不会对铁的氧化产生显著的影响。5 {. c; q' g* d e# R9 l; L
图12 Fe(100)、(110)、(111)在氧化1 ns时的侧视图[42] 1 a0 u& w: X9 `, k ~4 P; h
Fig. 12 Side-views of Fe (100), (110), (111) at 1 ns oxidation[42] 3.5 外电场电场是电荷及变化磁场周围空间里存在的一种特殊物质,电场由电荷和随时间变化的磁场产生,其会对处在场内的其他电荷产生力的作用。在微观尺度下,电场本质上是原子内原子核和电子之间的吸引力以及原子间的电子相互作用而形成的作用力[43]。反应力场分子动力学模拟在金属溶液模型之间引入一个外电场,在电场作用下,金属氧化膜的结构和形态会发生变化。一些分子动力学模拟研究表明,外电场会使本身不与水发生化学反应的金属发生腐蚀。Assowe等[25]建立了Ni(111)与纯水的金属溶液模型,镍的氧化膜厚度随电场强度的变化如图13所示。模拟结果表明随着电场强度的增加,氧化膜厚度呈线性增加趋势,镍的腐蚀速率加快。
+ I, i, v+ P4 y 图13 镍的氧化膜厚度随电场强度的变化[25] 1 E% p$ K0 z9 G, W6 M+ |/ I6 z% j9 n
Fig. 13 Variation of nickel oxide film thickness with electric field intensity[25] 0 m% ]) c, F! o
4 结 语 (1)国内外学者对金属材料腐蚀的反应力场分子动力学模拟进行了很多研究,从这些模拟结果来看,金属材料的腐蚀受到多种因素的影响,在分子/原子尺度上的腐蚀和宏观尺度上的腐蚀存在着明显的区别,使得到目前为止仍很难从本质上给出腐蚀的起源并归纳普适的腐蚀规律。(2)在未来腐蚀分子动力学模拟领域研究中,需要在以下几个方向开展重点研究:第一,加强对一些不完整金属氧化膜表面腐蚀机理的研究,例如金属的点缺陷、位错、晶界、杂质等缺陷模型[44]的研究,从原子角度阐述其腐蚀的机理。第二,建立能够反应真实宏观尺寸表面的金属腐蚀分子动力学模拟的模型,确定在什么参数下分子/原子尺度可以收敛到真实宏观尺度。第三,加强金属材料腐蚀分子动力学模拟结果与试验结果之间的相互关联和桥接,完善腐蚀理论[25]。
# ^0 A! f! p9 J0 L 参考文献:略
5 Z3 @' H, Z$ q* N2 e l- H 来源:材料保护
( P4 x% _' |0 ? X7 L% t: ]
RECOMMEND推荐阅读●收藏!362页PPT讲透防腐蚀工程技术●全名单下载!关于中国腐蚀与防护学会企业理事会●一种新型的周浸、浸润、循环腐蚀复合试验箱——CorrSky S100 型周浸腐蚀试验箱●2021第八届海洋材料与腐蚀防护大会在贵州隆重召开●企业风采:富钢集团致力于普惠超级钢筋的应用,服务全球超级工程建设●企业风采:宁波科鑫腐蚀控制工程有限公司——打造顶级防腐工程企业,争做行业领头雁 ●零声科技:为中国工业安全保驾护航中国腐蚀与防护网广告合作请联系:王 元 010-62316606-806或13693251529(微信同号)齐颖欣 010-62316606-801或18513781826(微信同号)
" A7 n3 }5 B$ y3 S, D
, F2 S) ?# Z- Q% U( k" Z5 }3 r) X5 s/ _; Q4 w7 q! M7 v1 T
|


