|
' j& d2 \7 Y7 L# R; M. k

$ W/ R2 e o; h" J& l
Extracting value: mechanistic insights into the formation of natural product artifacts – case studies in marine naturalproducts
4 O5 Q- E" `* U8 s 结构多样的活性天然产物使生物能够从容地应对环境变化,但一些活性产物可能源于人工转化,在提取、分离、保存等过程中哪怕微小的pH与温度的变化,氧气与光照的影响,以及与有机溶剂或色谱介质的反应,都可能导致人工产物的衍生。尽管如此,人工产物与天然产物间的关系与人工产物的形成机制仍然易被忽视。昆士兰大学Capon教授就此对海洋来源天然产物人工产物的形成机制与典型案例做了系统的分析和总结,本文节选其中溶液作用、pH效应及自发反应过程中的化学反应部分内容。 4 i# c. p6 j+ L$ z
溶液作用 " y! F% n" C! |/ {$ M
含有羧酸、环缩醛、环氨基、醌甲基及内酯等官能团或结构单元的化合物在醇类溶液中会衍生化生成人工产物;含有羧基的化合物在使用醇类溶液萃取或保存时会发生酯化反应,生成相应的人工产物。分离自海绵的carduusyne A(1)在乙醇中生成相应的乙酯2;分离自海洋真菌的flavipesolide C(4)在甲醇中生成甲酯flavipesolide B(5);分离自海绵的磷酸二酯聚酮类化合物franklinolide A(6)易在甲醇中酯化生成7,在氘代试剂CD3OD中酯化生成8。但这并不意味着乙酯化合物都是人工产物,如分离自海洋真菌的flavipesolide A(3)为天然乙酯类化合物;也并非所有人工酯化产物均可轻易辨出,如聚酮类didemnaketal B(9)最初报道为二甲酯天然产物,在发现其同系物didemnaketal C(10)在乙醇中长时间保存时酯基发生取代后,推测9中C-28酯基并非天然合成(Figure 1)。
; d; Y5 l# T, z' F1 P8 \) z 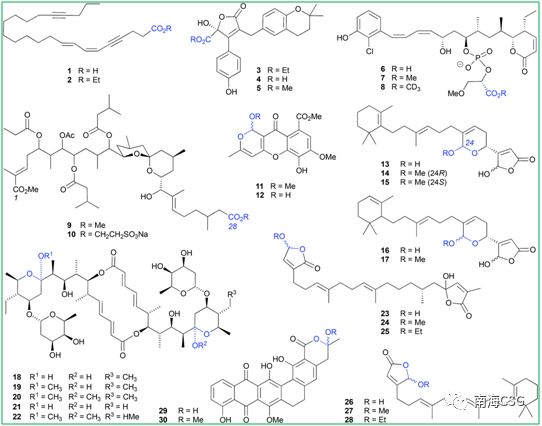
+ g% V6 ~: k2 Q2 x: C! |
Figure 1. 羧酸、环缩醛的醇溶剂分解举例
! _$ v" S: N8 L5 {' ~' E 含有环缩醛结构单元的化合物易在酸性条件下发生脱水,脱水中间体被醇加成后生成邻烷基缩醛(scheme 1),加成反应过程中若存在相邻手性中心的不对称诱导,则生成立体选择性手性化合物,反之,则生成一对差向异构体混合物。海洋藻类内生真菌来源的chaetocyclinone A(12)与MeOH加成生成邻甲基环缩醛chaetocyclinone A(11);海绵来源的cyclicacetal manoalide(13)与甲醇发生加成生成邻甲基14和15;海绵来源化合物16经硅胶柱色谱(MeOH/CHCl3)分离时生成luffariolide H(17);而洋橄榄叶素18与MeOH发生环缩醛反应立体选择性生成19和20;共代谢物efomycin G(21)生成11,11′-O-dimethyl-14′deethyl-14′-methylelaiophylin(22);二倍半萜cacolide C(23)加成形成人工化合物cacolides D(24)和E(25)。相似的反应也发生在海绵来源化合物luffariellolide(26)的MeOH/EtOH分离过程中,最后形成人工产物27和28(Figure 1)。 9 p8 h' A- _8 p' F
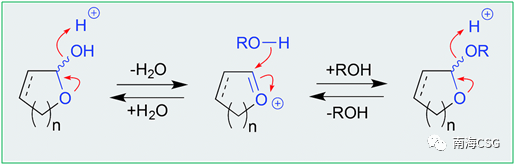
0 X/ h4 W! b6 r1 Q* l7 u3 T Scheme 1. 缩醛的醇溶剂分解机制
9 C1 L3 ]& A; c" h) p2 E pH效应 , W) Q2 z' {- D
天然产物在萃取、分离、保存过程中或多或少会接触酸性环境,天然产物本身含有酸性基团(如含酚羟基、羧基);次级代谢物中含酸性基团的化合物;一些有机试剂显弱酸性(CHCl3和DMSO);添加酸性试剂(色谱分离中添加微量的TFA);以及色谱相显酸性(硅胶)。这些酸性环境有时会导致天然产物发生反应生成人工产物。含有邻-异戊烯基的酚类(无论异戊烯基侧链是否氧化)在酸性条件下,萃取分离时均易发生环化反应(Scheme 2),因此应尽量避免在酸性环境下处理此类化合物,以减少人工产物的生成。遗憾的是,目前相关文献中还没有无酸对化合物影响的记载。135-144(Figure 2)是含有邻-异戊烯基的酚类化合物,136和137可由135环化或羟化形成;139和140可由138环化形成,而138本身可由135氧化生成;分离自海绵的141a/b是142的环化产物;没药烯倍半萜烯(+)-12-hydroxysydonic acid(143),分离自红藻真菌,在酸性条件下脱水环化形成144,这些环化或羟化可能都是萃取分离过程中自发发生的,文献没有指出这一点(Figure 2)。
5 y) W% ?" @! _+ { 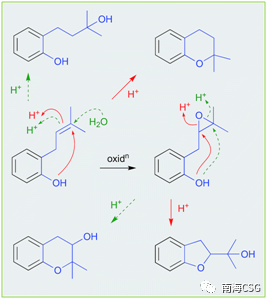
: ]0 a& h. Q" h( X Scheme 2. 酸性条件下,6/6环重排机制以及酚羟基重排机制
8 j ]) Q+ _$ ~* q# H 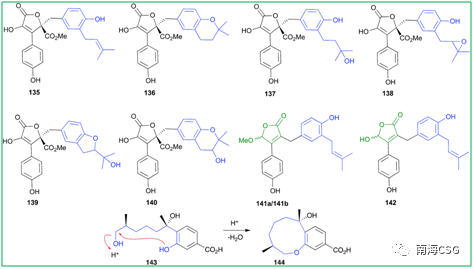
9 X: z3 c* ?* R/ M. R
Figure 2. 酸性条件下,分离过程中产生的非天然产物 5 T! k( ^' |7 x1 K% ?
化学反应
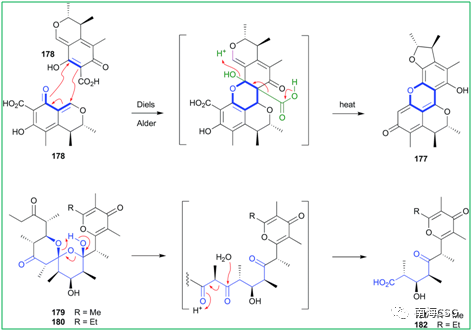
2 t. [4 ]! q7 D5 @, A 天然产物在自发形成人工产物的过程中会发生化学反应,包括DA-反应,逆克莱森缩合,迈克尔加成,席夫碱反应,苄基反应等。Citrinin(178)在MeOH溶液中,50℃条件下自发发生DA-反应,生成杂环DicitrininA(177),177此前被认为是海洋真菌与藻类共培养的天然产物;DihydrosiphonarinA(179)和B(180)是从Siphonaria spp.中分离得到的聚酮类天然产物,容易经过逆克莱森缩合转化为非天然产物181-182(Figure 3)。 ( `5 o4 z1 N( U/ J
9 u' K! c6 n- Y/ o: Y: T t! P
Figure 3. Diels-Alder和克莱森缩合反应产物
. W: j x1 M; u- Q% C/ {% G" O5 [ 聚酮化合物abyssomicin C(183)是氨基苯甲酸的生物合成抑制剂,183和184是一对差向异构体,在核磁共振数据采集过程中观察到184在CDCl3中不稳定,可转化为183;abyssomicin J(185)是183的类似物,推测185由两分子的183(或184)通过迈克尔加成反应二聚化生成,这个反应过程的解析促使仿生生物合成的成功;(10E, 15R)-10, 11-脱氢姜黄素(186)通过迈克尔加成反应生成187;Rhytidenone F(188)通过迈克尔加成形成rhytidenones C-E(189-191)(Figure 4)。
; R/ p/ w* r8 G) D [ 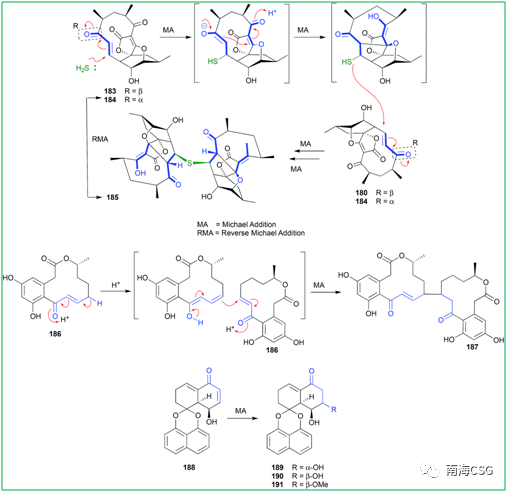
& y6 O6 [- W4 I% o Figure 4. Michael加成反应的人工产物
2 a u- }1 |1 f9 k, A 综上所述,本文节选了多种海洋来源天然产物因分离过程中溶液作用、pH效应发生化学反应生成人工产物的案例与机制。天然产物在转化为人工产物后活性往往发生变化,且人工产物的获得不利于天然产物合成途径的推导,因此在解析化合物结构后可根据其结构特征与文中的案例相比对,判断是否存在人工产物的可能。人工产物的形成大多无立体选择性,往往形成一对差向异构体混合物,据此,也可以初步判断获得的化合物是否为人工产物。对于易发生化学反应的化合物,如何更好地应用分离手段,采取有效措施避免人工产物的形成,除文中给我们的提示外,还需要更多的实践探索。 : b4 S: C! u/ G- ~' L8 D* F
本文作者为澳大利亚昆士兰大学分子生物学教授,主要研究海洋微生物天然产物,团队发现数千种天然产物,包括多种骨架新颖、结构复杂含多个官能团的生物活性小分子。 $ X' M. P; `) X' O" u
文章信息:Robert J. Capon; Extracting value: mechanistic insights into the formation of natural product artifacts – case studies in marine natural products; Nature Product Report, 2019, Advance Article.
. h+ w0 y: _7 F2 d 文章链接:https://pubs.rsc.org/en/content/articlelanding/2019/NP/C9NP00013E#!divAbstract
8 t2 q, i9 H. {! D0 k 文献阅读:王璐、叶伟霞、谭瑛(2019级硕士生) 7 ?; g) [: L" [
整理:张文军副研究员
7 B" x3 u0 `- G- Q M, B) [ 中国科学院南海海洋研究所 4 e" T4 k6 J, F/ o4 ^

2 Z! a0 [5 T8 ~' G0 J- G
& k" W0 c: O# R/ R8 O( P9 ?, i7 B6 W+ d; V) @0 s
| 

